Lowering your IOP may not be enough to stop your Glaucoma vision loss!
Neuroprotection, neurotrophins, ocular blood flow, ocular inflammation and oxidative stress can all contribute to your Glaucoma progression.
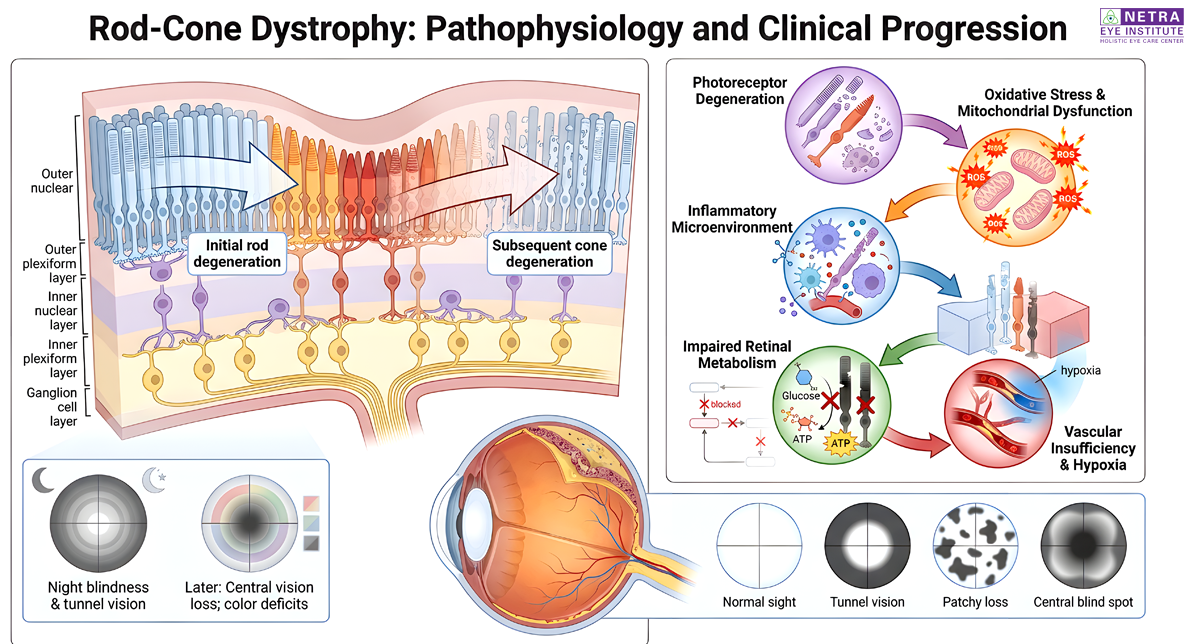
Retinitis Pigmentosa is a group of rare genetic disorders that affect the retina's ability to respond to light. This condition typically begins with a loss of night vision and a narrowing of the visual field, often called tunnel vision. While conventional medicine focuses on management, a holistic Retinitis Pigmentosa Treatment looks at ways to support the remaining retinal cells. At our institute, we prioritize natural methods to slow the progression and maintain visual function.
The Role of Ayurvedic Eye Treatment
In the realm of traditional healing, retinal health depends on the balance of internal nutrients and energy. An Ayurvedic eye treatment for this condition involves the use of specialized herbs to nourish the photoreceptor cells. This specific retinitis pigmentosa treatment aims to reduce oxidative stress within the eye, which is a major factor in cell decay. These time-tested practices help provide the eye with a supportive environment for better stability.
Benefits of Integrative Eye Care
Managing a genetic condition requires a multi-faceted strategy that looks at the body as a whole. Our integrative eye care approach combines Traditional Chinese Medicine and Acupuncture with herbal protocols. This comprehensive retinitis pigmentosa treatment focuses on improving ocular blood flow and metabolic health. By merging these ancient disciplines, we offer a path that differs from standard practices by focusing on long-term cellular support rather than just symptom management.
Natural Recovery and Retinal Support
Protecting your remaining sight involves a commitment to whole-body wellness. A successful retinitis pigmentosa treatment uses antioxidants and herbal medicines to help the retina function at its best. By using integrative eye care, we help patients find natural ways to manage their condition. Our ayurvedic eye treatment options provide a clear distinction for those seeking a gentle and effective way to support their ocular health. Our focus remains on your ability to maintain a high quality of life through better vision.
Neuroprotection, neurotrophins, ocular blood flow, ocular inflammation and oxidative stress can all contribute to your Glaucoma progression.
Glaucoma is a neurodegenerative disease, not just a pressure disease
Glaucoma is increasingly understood as a neurodegenerative disease, not merely a condition caused by elevated eye pressure. Mechanisms such as impaired ocular blood flow, mitochondrial dysfunction, oxidative stress, chronic inflammation, and reduced neurotrophic support, all of which contribute to neuronal injury.
Optic nerve damage isn't always caused by high eye pressure
While elevated intraocular pressure (IOP) is a major risk factor, many people develop glaucoma even with normal or low eye pressure—a condition known as normal-tension glaucoma (NTG). Recognizing glaucoma as a neurodegenerative condition underscores the need for treatment strategies that go beyond pressure control and actively support optic nerve health and neuroprotection.
Lowering IOP is important, but not always enough
Studies show that 30–40% of glaucoma patients continue to lose vision despite maintaining normal eye pressure. This highlights the need for a more comprehensive glaucoma treatment approach that goes beyond just IOP management.
Individualized care is essential
These patients may require more than just IOP-lowering treatments. For instance, neuroprotective strategies targeting the optic nerve or treatments to improve ocular blood flow to the retina and optic nerve may be considered to reduce progression.
What causes continued vision loss in glaucoma?
Multiple research studies have shown factors such as lack of neuroprotection, reduced ocular blood flow, neurotrophin deprivation, increased oxidative stress, ocular inflammation and excitotoxicity can all contribute to vision loss progression in Glaucoma.
Lowering intraocular pressure (IOP) with eye drops has long been the standard approach in glaucoma care. However, IOP control alone does not fully address the disease process. Many patients continue to experience optic nerve damage and vision loss despite well-controlled eye pressure, highlighting the need for a more comprehensive treatment strategy that supports optic nerve health and addresses underlying factors beyond pressure alone.

Normal Tension Glaucoma (NTG) is a type of glaucoma in which optic nerve damage and vision loss occur despite eye pressure remaining within the normal range. In NTG, pressure alone does not explain the disease; instead, the optic nerve is thought to be more vulnerable due to factors such as reduced blood flow, impaired circulation regulation, increased nerve sensitivity, oxidative stress, and reduced neuroprotection. Because of this heightened susceptibility, even normal eye pressure can contribute to progressive damage over time.
The mean percentage of NTG in all patients diagnosed with a glaucomatous visual field defect is between 30% and 40%.
NTG is especially common in Asian populations, where it can account for a majority of primary open-angle glaucoma (POAG) cases—often exceeding 70–90%. This highlights that glaucoma is not solely a pressure-related condition, but a complex disease influenced by overall optic nerve health and systemic factors.
The prevalence of NTG has been reported to be 92.3% in Japan, 84.6% in Singapore, 83.58% in Northern China, 82% in South India, 79.3% in Southern China, 77% in South Korea, 57.1% in South Africa, 46.9% in Iran, 38.9% in Netherlands, 31.7% in the United States, 31.0% in Iceland, and 30.0% in Italy.
Normal Tension Glaucoma (NTG) patients often requires more than just eye-pressure–lowering drops because eye pressure is not their main cause of glaucomatous vision loss. In NTG, vision loss can occur even when eye pressure is normal, due to reduced blood flow to the optic nerve, poor regulation of blood vessels, increased inflammation, oxidative stress, and reduced support for the nerve cells that carry visual signals to the brain. While eye drops may help lower pressure slightly and remain part of treatment, they do not improve optic nerve blood flow or protect nerve cells from ongoing injury. For this reason, NTG care often needs a broader approach that supports optic nerve health, stabilizes blood flow, and addresses the underlying factors contributing to nerve damage—beyond eye pressure alone.
Our signature Netra Restoration Therapy is a unique treatment method available exclusively at Netra Eye Institute, which has shown to halt AMD progression, improve visual acuity, reduce foggy/hazy vision, improve contrast sensitivity and reduce glare.
The Mechanism of Action (MOA) of Netra Restoration Therapy works by enhancing ocular blood flow through the regulation of vascular function, reducing oxidative stress and ocular inflammation, increasing neurotrophin levels and neuroprotection, and reducing ferroptosis.
The potential for visual improvement depends on the severity of retinal damage present at the time of treatment for Wet AMD. Taking these factors into account, our therapeutic approach has been shown to result in:

Stops vision loss progression by reducing the ocular inflammation, regulating ocular blood flow and nourshing the retinal cells.
Improvement in visual field by restoring
dormant and unhealthy retinal cells.
An improvement of at least one line on the distance and near vision eye chart.
Improved contrast vision, making it easier to distinguish shapes, edges, and details.
Improvement in color, brightness perception and clarity making it easier to see in low-light or nighttime conditions, thereby supporting safer mobility and daily activities.
Reduced glare, less light sensitivity, and improved comfort in bright environments, such as sunlight, headlights, or digital screens.
Patients experience considerable reduction
in eye pain and eye strains.
Patients often report feeling “healthier overall,” not just in their eyes.

I recently completed my three weeks of Netra Restoration Therapy and I am already seeing major improvements with the reduction in my eye prescription correction numbers. Netra Eye Institute is not just a well equipped clinic with modern ophthalmic instruments but is also backed up by professional and patient-caring staff. Thank you Dr. Gandapodi for your services and care.
– Sarang - Grateful Netra Patient
Netra Restoration Therapy is grounded in contemporary biomedical research demonstrating that many eye diseases are driven by reduced ocular blood flow, ongoing neurodegeneration, and cellular stress. Scientific studies show that improving vascular regulation enhances oxygen and nutrient delivery to the retina and optic nerve, while supporting neurotrophin activity and neuroprotection helps preserve vulnerable nerve cells. At the same time, controlling oxidative stress, ferroptosis, and chronic inflammation is critical to slowing tissue damage and disease progression. Netra Restoration integrates these evidence-based principles into a comprehensive approach designed to support long-term eye health and visual function.






Want to learn more about Netra Restoration Therapy and holistic eye treatments or need help to schedule an appointment for a consultation?
Call us at (732)-503-9999 or fill out the contact us form below.



.webp)